Combine 2 pathfindR Results
combine_pathfindR_results(result_A, result_B, plot_common = TRUE)Arguments
Value
Data frame of combined pathfindR enrichment results. Columns are:
- ID
ID of the enriched term
- Term_Description
Description of the enriched term
- Fold_Enrichment_A
Fold enrichment value for the enriched term (Calculated using ONLY the input genes)
- occurrence_A
the number of iterations that the given term was found to enriched over all iterations
- lowest_p_A
the lowest adjusted-p value of the given term over all iterations
- highest_p_A
the highest adjusted-p value of the given term over all iterations
- Up_regulated_A
the up-regulated genes in the input involved in the given term's gene set, comma-separated
- Down_regulated_A
the down-regulated genes in the input involved in the given term's gene set, comma-separated
- Fold_Enrichment_B
Fold enrichment value for the enriched term (Calculated using ONLY the input genes)
- occurrence_B
the number of iterations that the given term was found to enriched over all iterations
- lowest_p_B
the lowest adjusted-p value of the given term over all iterations
- highest_p_B
the highest adjusted-p value of the given term over all iterations
- Up_regulated_B
the up-regulated genes in the input involved in the given term's gene set, comma-separated
- Down_regulated_B
the down-regulated genes in the input involved in the given term's gene set, comma-separated
- combined_p
the combined p value (via Fisher's method)
- status
whether the term is found in both analyses ('common'), found only in the first ('A only') or found only in the second ('B only)
By default, the function also displays the term-gene graph of the common terms
Examples
combined_results <- combine_pathfindR_results(example_pathfindR_output, example_comparison_output)
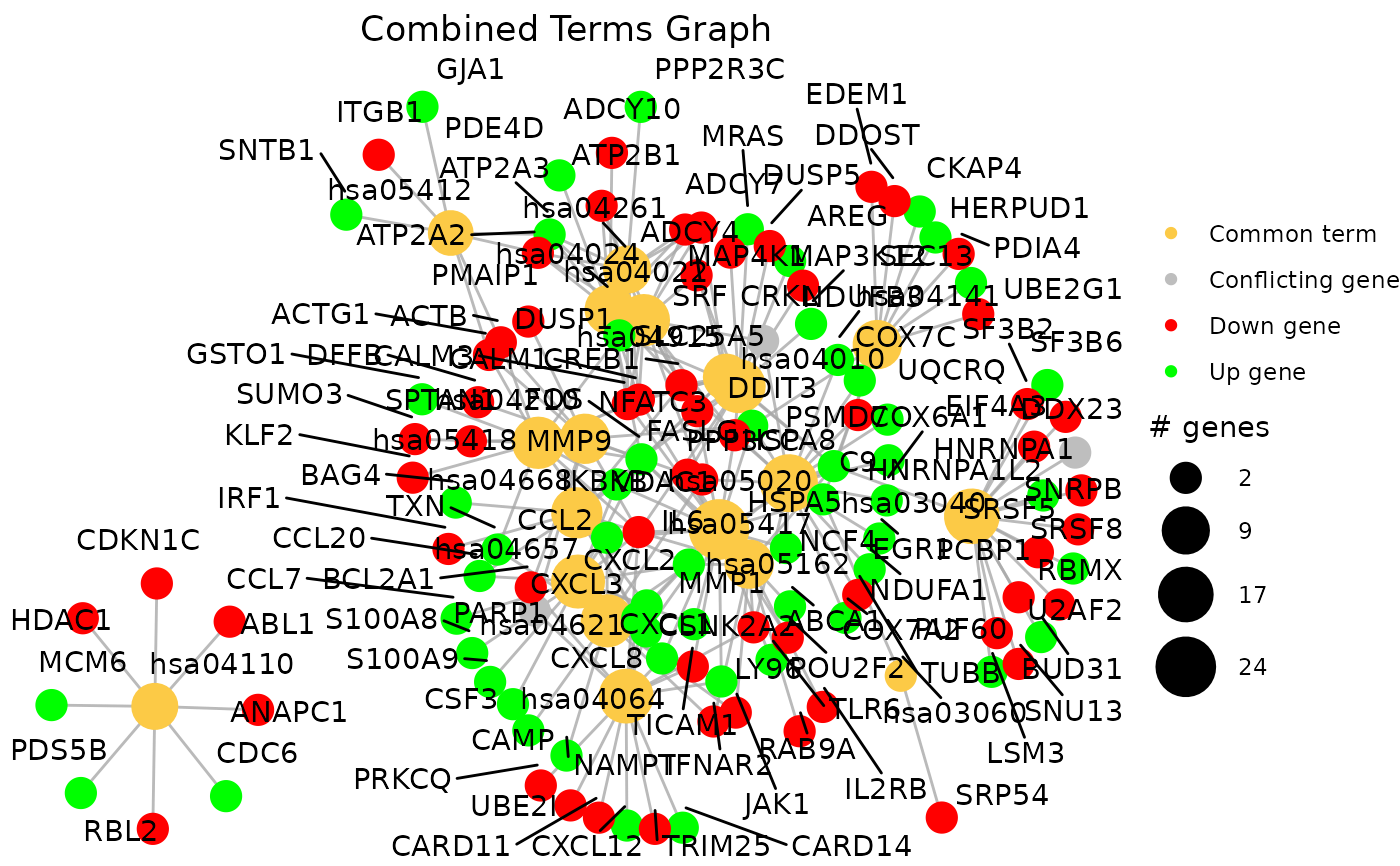 #> You may run `combined_results_graph()` to create visualizations of combined term-gene graphs of selected terms
#> You may run `combined_results_graph()` to create visualizations of combined term-gene graphs of selected terms