Introduction to pathfindR
Ege Ulgen
2026-07-02
Source:vignettes/intro_vignette.Rmd
intro_vignette.RmdpathfindR is a tool for enrichment analysis via active
subnetworks. The package also offers functionality to cluster the
enriched terms and identify representative terms in each cluster, to
score the enriched terms per sample and to visualize analysis
results.
The functionality suite of pathfindR is described in detail in Ulgen E, Ozisik O, Sezerman OU. 2019. pathfindR: An R Package for Comprehensive Identification of Enriched Pathways in Omics Data Through Active Subnetworks. Front. Genet. https://doi.org/10.3389/fgene.2019.00858
Overview
The observation that motivated us to develop pathfindR
was that direct enrichment analysis of differential RNA/protein
expression or DNA methylation results may not provide the researcher
with the full picture. That is to say: enrichment analysis of only a
list of significant genes alone may not be informative enough to explain
the underlying disease mechanisms. Therefore, we considered leveraging
interaction information from a protein-protein interaction network (PIN)
to identify distinct active subnetworks and then perform enrichment
analyses on these subnetworks.
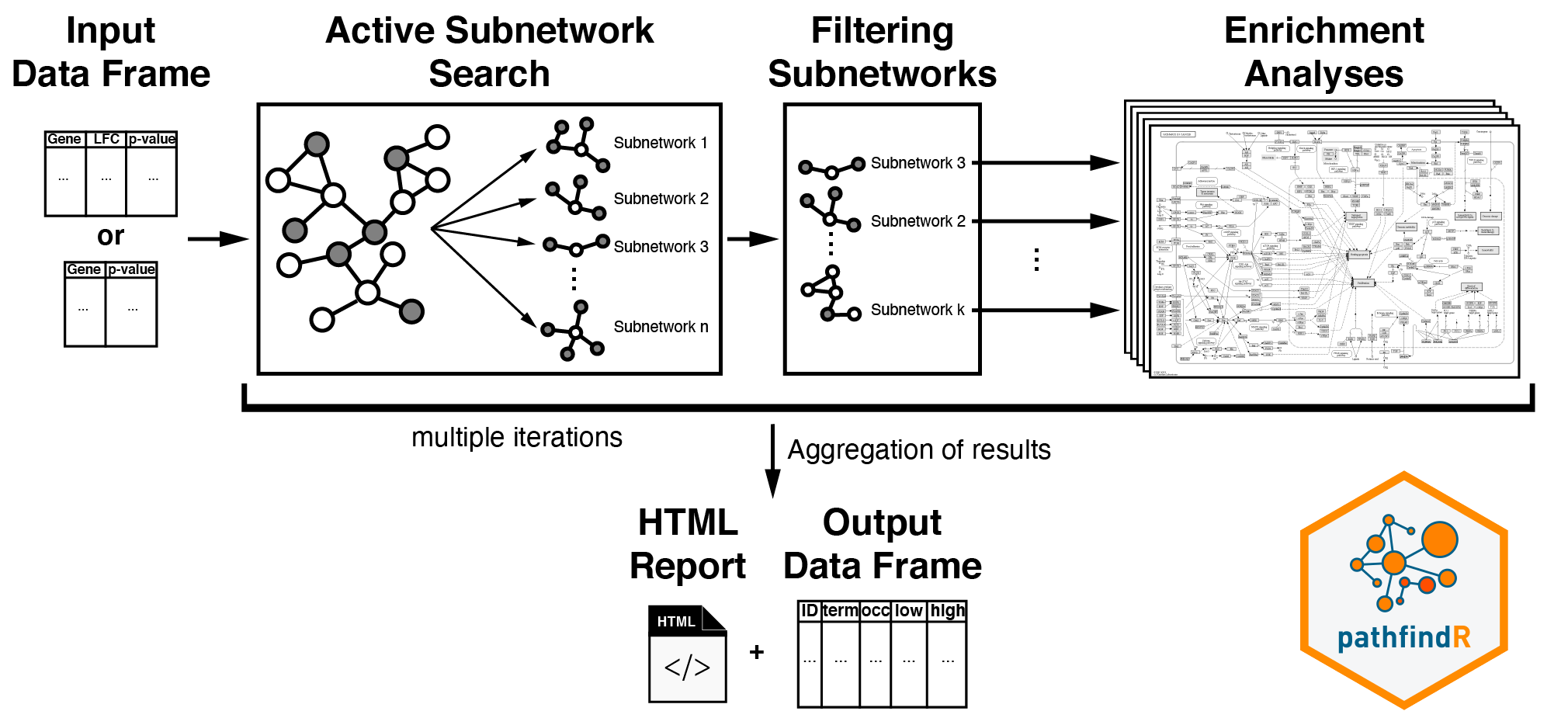
An active subnetwork can be defined as a group of interconnected genes in a PIN that predominantly consists of significantly altered genes. In other words, active subnetworks define distinct disease-associated sets of interacting genes, whether discovered through the original analysis or discovered because of being in interaction with a significant gene.
The active-subnetwork-oriented enrichment analysis approach of
pathfindR can be summarized as follows: Mapping the input genes with the
associated p values onto the PIN (after processing the input), active
subnetwork search is performed. The resulting active subnetworks are
then filtered based on their scores and the number of significant genes
they contain. This filtered list of active subnetworks are then used for
enrichment analyses, i.e. using the genes in each of the active
subnetworks, the significantly enriched terms (pathways/gene sets) are
identified. Enriched terms with adjusted p values larger than the given
threshold are discarded and the lowest adjusted p value (over all active
subnetworks) for each term is kept. This process of
active subnetwork search + enrichment analyses is repeated
for a selected number of iterations, performed in parallel. Over all
iterations, the lowest and the highest adjusted-p values, as well as
number of occurrences over all iterations are reported for each
significantly enriched term in the resulting data frame. This
active-subnetwork-oriented enrichment approach is demonstrated in the
section Active-subnetwork-oriented
Enrichment Analysis of this vignette.
The enrichment analysis usually yields a great number of enriched terms whose biological functions are related. Therefore, we implemented two clustering approaches using a pairwise distance matrix based on the kappa statistics between the enriched terms (as proposed by Huang et al. 1). Based on this distance metric, the user can perform either hierarchical (default) or fuzzy clustering of the enriched terms. Details of clustering and partitioning of enriched terms are presented in the Clustering Enriched Terms section of this vignette.
Other functionality of pathfindR includes:
- agglomerated score calculation per each term (to investigate how a gene set is altered in a given sample)
- visualization of terms and term-related genes as a graph (to determine the degree of overlap between the enriched terms by identifying shared and/or distinct significant genes) and
- pathfindR analysis with custom gene sets are also briefly described.
Active-subnetwork-oriented Enrichment Analysis
For convenience, we provide the wrapper function
run_pathfindR() to be used for the
active-subnetwork-oriented enrichment analysis. The input for this
function must be a data frame consisting of the columns containing:
Gene Symbols, Change Values (optional) and
p values. The example data frame used in this vignette
(example_pathfindR_input) is the dataset containing the
differentially-expressed genes for the GEO dataset GSE15573 comparing 18
rheumatoid arthritis (RA) patients versus 15 healthy subjects.
The first 6 rows of the example input data frame are displayed below:
| Gene.symbol | logFC | adj.P.Val |
|---|---|---|
| FAM110A | -0.6939359 | 0.0000034 |
| RNASE2 | 1.3535040 | 0.0000101 |
| S100A8 | 1.5448338 | 0.0000347 |
| S100A9 | 1.0280904 | 0.0002263 |
| TEX261 | -0.3235994 | 0.0002263 |
| ARHGAP17 | -0.6919330 | 0.0002708 |
Executing the workflow is straightforward (but does typically take several minutes):
output_df <- run_pathfindR(example_pathfindR_input)Useful arguments
This subsection demonstrates some (selected) useful arguments of
run_pathfindR(). For a full list of arguments, see
?run_pathfindR or visit our GitHub
wiki.
Filtering Input Genes
By default, run_pathfindR() uses the input genes with
p-values < 0.05. To change this threshold, use
p_val_threshold:
output_df <- run_pathfindR(example_pathfindR_input, p_val_threshold = 0.01)Output Directory
By default, run_pathfindR() creates a temporary
directory for writing the output files, including active subnetwork
search results and a HTML report. To set the output directory, use
output_dir:
output_df <- run_pathfindR(example_pathfindR_input, output_dir = "this_is_my_output_directory")This creates "this_is_my_output_directory" under the
current working directory.
In essence, this argument is treated as a path so it can be used to
create the output directory anywhere. For example, to create the
directory "my_dir" under "~/Desktop" and run
the analysis there, you may run:
output_df <- run_pathfindR(example_pathfindR_input, output_dir = "~/Desktop/my_dir")Note: If the output directory (e.g.
"my_dir") already exists,run_pathfindR()creates and works under"my_dir(1)". If that exists also exists, it creates"my_dir(2)"and so on. This was intentionally implemented so that any previous pathfindR results are not overwritten.
Gene Sets for Enrichment
The active-subnetwork-oriented enrichment analyses can be performed
on any gene sets (biological pathways, gene ontology terms,
transcription factor target genes, miRNA target genes etc.). The
available gene sets in pathfindR are “KEGG”, “Reactome”, “BioCarta”,
“GO-All”, “GO-BP”, “GO-CC” and “GO-MF” (all for Homo sapiens). For
changing the default gene sets for enrichment analysis (hsa KEGG
pathways), use the argument gene_sets
output_df <- run_pathfindR(example_pathfindR_input, gene_sets = "GO-MF")By default, run_pathfindR() filters the gene sets by
including only the terms containing at least 10 and at most 300 genes.
To change the default behavior, you may change
min_gset_size and max_gset_size:
## Including more terms for enrichment analysis
output_df <- run_pathfindR(example_pathfindR_input,
gene_sets = "GO-MF",
min_gset_size = 5,
max_gset_size = 500
)Note that increasing the number of terms for enrichment analysis may result in significantly longer run time.
If the user prefers to use another gene set source, the
gene_sets argument should be set to "Custom"
and the custom gene sets (list) and the custom gene set descriptions
(named vector) should be supplied via the arguments
custom_genes and custom_descriptions,
respectively. See ?fetch_gene_sets for more details and Analysis with Custom Gene
Sets for a simple demonstration.
For details on obtaining organism-specific Gene Sets and PIN data, see the vignette Obtaining PIN and Gene Sets Data.
Filtering Enriched Terms by Adjusted-p Values
By default, run_pathfindR() adjusts the enrichment p
values via the “bonferroni” method and filters the enriched terms by
adjusted-p value < 0.05. To change this adjustment method and the
threshold, set adj_method and
enrichment_threshold, respectively:
output_df <- run_pathfindR(example_pathfindR_input,
adj_method = "fdr",
enrichment_threshold = 0.01
)Protein-protein Interaction Network
For the active subnetwork search process, a protein-protein
interaction network (PIN) is used. run_pathfindR() maps the
input genes onto this PIN and identifies active subnetworks which are
then be used for enrichment analyses. To change the default PIN
(“Biogrid”), use the pin_name_path argument:
output_df <- run_pathfindR(example_pathfindR_input, pin_name_path = "IntAct")The pin_name_path argument can be one of “Biogrid”,
“STRING”, “GeneMania”, “IntAct”, “KEGG”, “mmu_STRING” or it can be the
path to a custom PIN file provided by the user.
# to use an external PIN of your choice
output_df <- run_pathfindR(example_pathfindR_input, pin_name_path = "/path/to/myPIN.sif")NOTE: the PIN is also used for generating the background genes (in this case, all unique genes in the PIN) during hypergeometric-distribution-based tests in enrichment analyses. Therefore, a large PIN will generally result in better results.
Active Subnetwork Search Method
Currently, there are three algorithms implemented in pathfindR for active subnetwork search: Greedy Algorithm (default, based on Ideker et al. 2), Simulated Annealing Algorithm (based on Ideker et al. 3) and Genetic Algorithm (based on Ozisik et al. 4). For a detailed discussion on which algorithm to use see this wiki entry
# for simulated annealing:
output_df <- run_pathfindR(example_pathfindR_input, search_method = "SA")
# for genetic algorithm:
output_df <- run_pathfindR(example_pathfindR_input, search_method = "GA")Other Arguments
Because the active subnetwork search algorithms are stochastic,
run_pathfindR() may be set to iterate the active subnetwork
identification and enrichment steps multiple times (by default 1 time).
To change this number, set iterations:
output_df <- run_pathfindR(example_pathfindR_input, iterations = 25)run_pathfindR() uses a parallel loop (using the package
foreach) for performing these iterations in parallel. By
default, the number of processes to be used is determined automatically.
To override, change n_processes:
# if not set, `n_processes` defaults to (number of detected cores - 1)
output_df <- run_pathfindR(example_pathfindR_input, iterations = 5, n_processes = 2)Output
Enriched Terms Data Frame
run_pathfindR() returns a data frame of enriched terms.
Columns are:
- ID: ID of the enriched term
- Term_Description: Description of the enriched term
- Fold_Enrichment: Fold enrichment value for the enriched term (Calculated using ONLY the input genes)
- occurrence: The number of iterations that the given term was found to enriched over all iterations
- lowest_p: the lowest adjusted-p value of the given term over all iterations
- highest_p: the highest adjusted-p value of the given term over all iterations
- non_Signif_Snw_Genes (OPTIONAL): the non-significant active
subnetwork genes, comma-separated (controlled by
list_active_snw_genes, default isFALSE) - Up_regulated: the up-regulated genes (as determined by
change value> 0, if thechange columnwas provided) in the input involved in the given term’s gene set, comma-separated. If change column was not provided, all affected input genes are listed here. - Down_regulated: the down-regulated genes (as determined by
change value< 0, if thechange columnwas provided) in the input involved in the given term’s gene set, comma-separated
The first 2 rows of the output data frame of the example analysis on
the rheumatoid arthritis gene-level differential expression input data
(example_pathfindR_input) is shown below:
| ID | Term_Description | Fold_Enrichment | occurrence | support | lowest_p | highest_p | Up_regulated | Down_regulated |
|---|---|---|---|---|---|---|---|---|
| hsa05415 | Diabetic cardiomyopathy | 3.744993 | 10 | 0.0871034 | 0 | 0.000000 | NCF4, NDUFA1, NDUFB3, UQCRQ, COX6A1, COX7A2, COX7C, MMP9, GAPDH | SLC25A5, VDAC1, ATP2A2, MTOR, PDHA1, PDHB, PARP1 |
| hsa03082 | ATP-dependent chromatin remodeling | 4.213117 | 10 | 0.0245786 | 0 | 0.000312 | ARID1A, SMARCA4, ACTB, ACTG1, BRD7, BRD9, HDAC1, EP400, TRRAP |
By default, run_pathfindR() also produces a graphical
summary of enrichment results for top 10 enriched terms, which can also
be later produced by enrichment_chart():
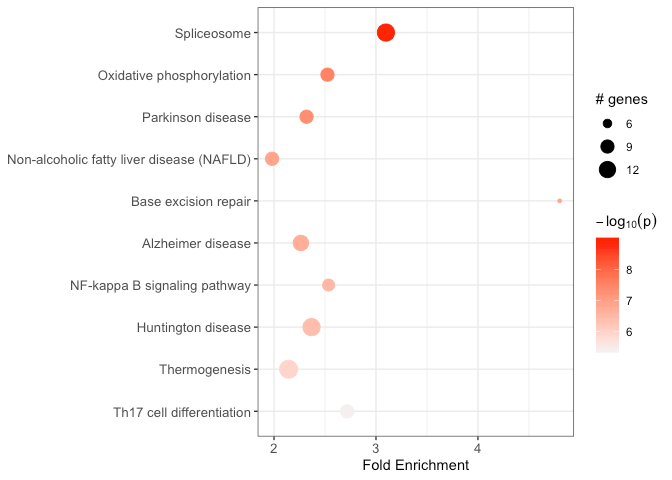
You may also disable plotting this chart by setting
plot_enrichment_chart=FALSE and later produce this plot via
the function enrichment_chart():
# change number of top terms plotted (default = 10)
enrichment_chart(
result_df = example_pathfindR_output,
top_terms = 15
)HTML Report (created when output_dir is set)
The function also creates an HTML report results.html
that is saved in the output directory if it’s set. This report contains
links to two other HTML files:
1. enriched_terms.html
This document contains the table of the active subnetwork-oriented enrichment results (same as the returned data frame).
2. conversion_table.html
This document contains the table of converted gene symbols. Columns are:
- Old Symbol: the original gene symbol
- Converted Symbol: the alias symbol that was found in the PIN
- Change: the provided change value
- p-value: the provided adjusted p value
During input processing, gene symbols that are not in the PIN are identified and excluded. For human genes, if aliases of these missing gene symbols are found in the PIN, these symbols are converted to the corresponding aliases (controlled by the argument
convert2alias). This step is performed to best map the input data onto the PIN.
The document contains a second table of genes for which no interactions were identified after checking for alias symbols (so these could not be used during the analysis).
Enriched Term Diagrams
For KEGG enrichment analyses, visualize_terms() can be
used to generate KEGG pathway diagrams that are returned as a list of
ggraph objects (using ggkegg)::
input_processed <- input_processing(example_pathfindR_input)
gg_list <- visualize_terms(
result_df = example_pathfindR_output,
input_processed = input_processed,
is_KEGG_result = TRUE
) # this function returns a list of ggraph objects (named by Term ID)
# save one of the plots as PDF image
ggplot2::ggsave(
"hsa04911_diagram.pdf",
gg_list$hsa04911,
width = 5,
height = 5
)Alternatively (i.e., for other types of non-KEGG enrichment
analyses), an interaction diagram per enriched term can be generated
again via visualize_terms(). These diagrams are also
returned as a list of ggraph objects:
input_processed <- input_processing(example_pathfindR_input)
gg_list <- visualize_terms(
result_df = example_pathfindR_output,
input_processed = input_processed,
is_KEGG_result = FALSE,
pin_name_path = "Biogrid"
) # this function returns a list of ggraph objects (named by Term ID)
# save one of the plots as PDF image
ggplot2::ggsave(
"diabetic_cardiomyopathy_interactions.pdf",
gg_list$hsa04911,
width = 10,
height = 6
)Clustering Enriched Terms
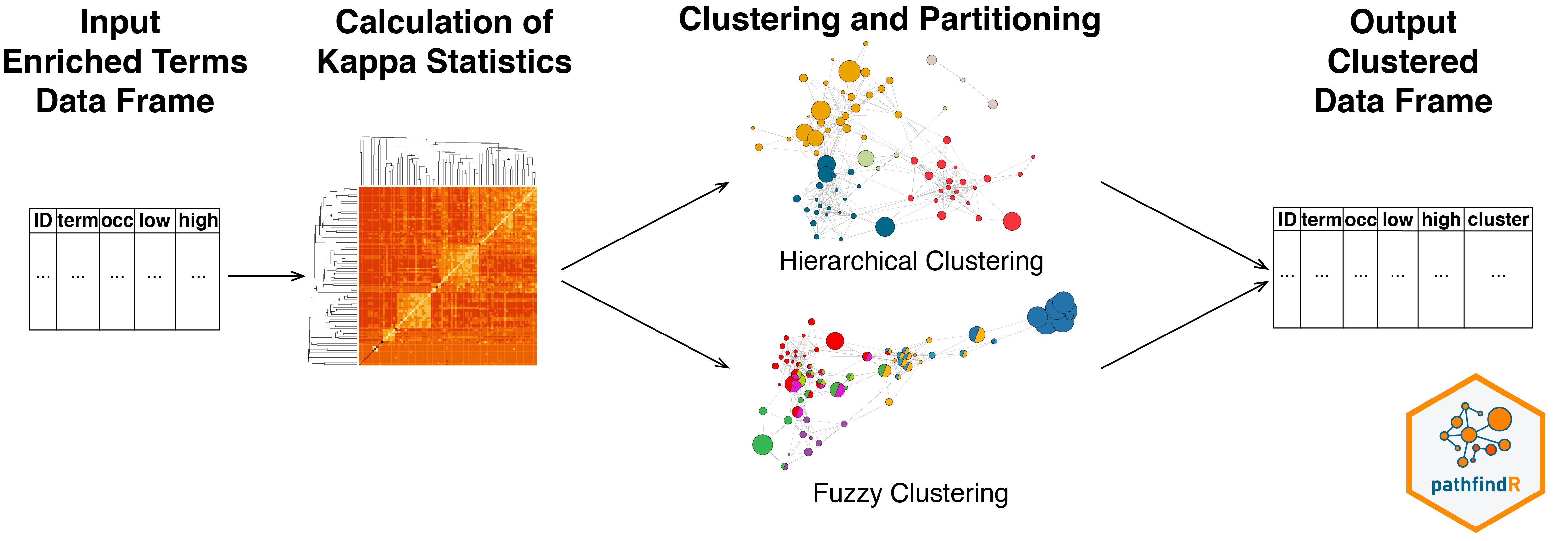
The wrapper function cluster_enriched_terms() can be
used to perform clustering of enriched terms and partitioning the terms
into biologically-relevant groups. Clustering can be performed either
via hierarchical or fuzzy method using the
pairwise kappa statistics (a chance-corrected measure of co-occurrence
between two sets of categorized data) matrix between all enriched
terms.
Hierarchical Clustering
By default, cluster_enriched_terms() performs
hierarchical clustering of the terms (using
as the distance metric). Iterating over
clusters (where
is the number of terms), cluster_enriched_terms()
determines the optimal number of clusters by maximizing the average
silhouette width, partitions the data into this optimal number of
clusters and returns a data frame with cluster assignments.
example_pathfindR_output_clustered <- cluster_enriched_terms(example_pathfindR_output, plot_dend = FALSE, plot_clusters_graph = FALSE)| ID | Term_Description | Fold_Enrichment | occurrence | support | lowest_p | highest_p | Up_regulated | Down_regulated | Cluster | Status |
|---|---|---|---|---|---|---|---|---|---|---|
| hsa05415 | Diabetic cardiomyopathy | 3.744993 | 10 | 0.0871034 | 0 | 0.000000 | NCF4, NDUFA1, NDUFB3, UQCRQ, COX6A1, COX7A2, COX7C, MMP9, GAPDH | SLC25A5, VDAC1, ATP2A2, MTOR, PDHA1, PDHB, PARP1 | 1 | Representative |
| hsa03082 | ATP-dependent chromatin remodeling | 4.213117 | 10 | 0.0245786 | 0 | 0.000312 | ARID1A, SMARCA4, ACTB, ACTG1, BRD7, BRD9, HDAC1, EP400, TRRAP | 1 | Member |
## The representative terms
knitr::kable(example_pathfindR_output_clustered[example_pathfindR_output_clustered$Status == "Representative", ])| ID | Term_Description | Fold_Enrichment | occurrence | support | lowest_p | highest_p | Up_regulated | Down_regulated | Cluster | Status | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | hsa05415 | Diabetic cardiomyopathy | 3.744993 | 10 | 0.0871034 | 0.0000000 | 0.0000000 | NCF4, NDUFA1, NDUFB3, UQCRQ, COX6A1, COX7A2, COX7C, MMP9, GAPDH | SLC25A5, VDAC1, ATP2A2, MTOR, PDHA1, PDHB, PARP1 | 1 | Representative |
| 3 | hsa04130 | SNARE interactions in vesicular transport | 4.936581 | 10 | 0.0338571 | 0.0000000 | 0.0000000 | STX6 | BET1L, SNAP23, STX2 | 2 | Representative |
| 7 | hsa03013 | Nucleocytoplasmic transport | 3.954058 | 10 | 0.0150067 | 0.0000000 | 0.0109558 | NUP214 | NUP62, NUP93, UBE2I, RANGAP1, SUMO3, EIF4A3, RNPS1, SRRM1, TNPO1 | 3 | Representative |
| 12 | hsa03083 | Polycomb repressive complex | 2.483341 | 10 | 0.0079062 | 0.0000007 | 0.0495676 | CBX3 | PHF19, RING1, KDM2B, HDAC1 | 4 | Representative |
| 14 | hsa00071 | Fatty acid degradation | 2.841404 | 1 | 0.0062893 | 0.0000034 | 0.0000034 | HADH, ECHS1, ALDH9A1 | 5 | Representative | |
| 18 | hsa03430 | Mismatch repair | 5.312191 | 10 | 0.0232130 | 0.0000088 | 0.0002342 | MLH1, POLD2, RPA1 | 6 | Representative | |
| 23 | hsa04141 | Protein processing in endoplasmic reticulum | 1.480974 | 7 | 0.0065359 | 0.0000170 | 0.0153838 | DDIT3, CKAP4 | DDOST, EDEM1, UBE2G1, PDIA4 | 7 | Representative |
| 46 | hsa03050 | Proteasome | 1.810080 | 10 | 0.0180547 | 0.0006553 | 0.0025872 | PSMD7, PSMB10 | 8 | Representative | |
| 124 | hsa03010 | Ribosome | 1.770730 | 1 | 0.0063694 | 0.0279007 | 0.0279007 | RPL26, RPL39, RPL31, MRPL33, MRPS18C, RPS24 | MRPL49, RPLP2 | 9 | Representative |
| 134 | hsa00052 | Galactose metabolism | 1.357560 | 4 | 0.0075034 | 0.0426637 | 0.0426637 | HK3 | 10 | Representative |
After clustering, you may again plot the summary enrichment chart and display the enriched terms by clusters:
# plotting only selected clusters for better visualization
selected_clusters <- subset(example_pathfindR_output_clustered, Cluster %in% 5:7)
enrichment_chart(selected_clusters, plot_by_cluster = TRUE)
#> Plotting the enrichment bubble chart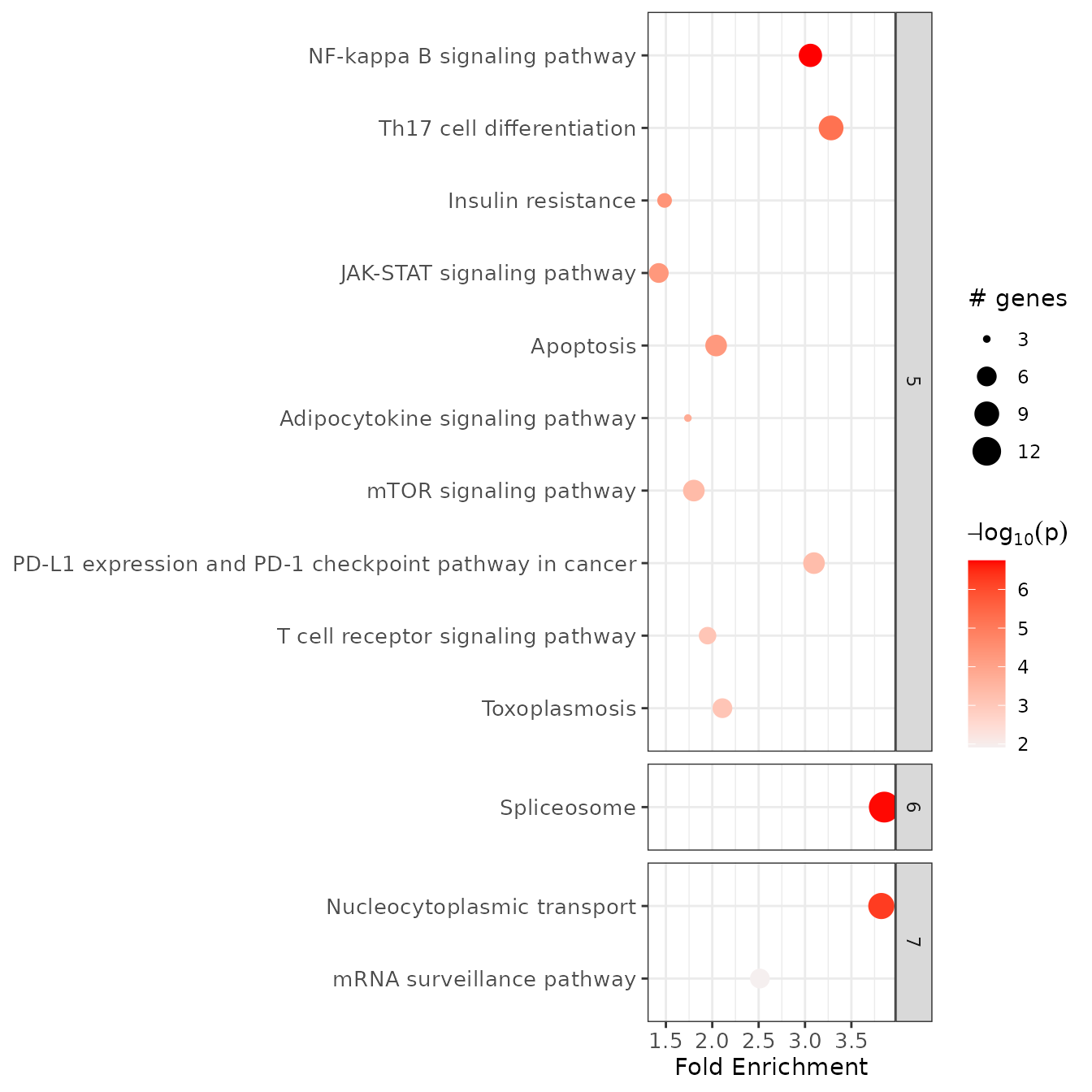
For details, see ?hierarchical_term_clustering
Heuristic Fuzzy Multiple-linkage Partitioning
Alternatively, the fuzzy clustering method (as described
by Huang et al.5) can be used:
clustered_fuzzy <- cluster_enriched_terms(example_pathfindR_output, method = "fuzzy")For details, see ?fuzzy_term_clustering
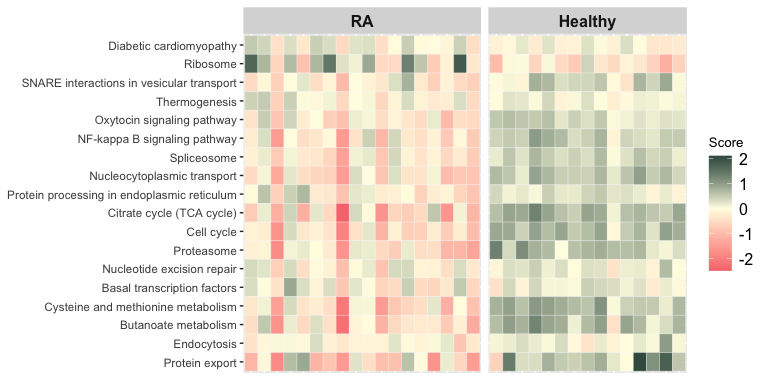
Aggregated Term Scores per Sample
The function score_terms() can be used to calculate the
agglomerated z score of each enriched term per sample. This allows the
user to individually examine the scores and infer how a term is overall
altered (activated or repressed) in a given sample or a group of
samples.
## Vector of "Case" IDs
cases <- c(
"GSM389703", "GSM389704", "GSM389706", "GSM389708",
"GSM389711", "GSM389714", "GSM389716", "GSM389717",
"GSM389719", "GSM389721", "GSM389722", "GSM389724",
"GSM389726", "GSM389727", "GSM389730", "GSM389731",
"GSM389733", "GSM389735"
)
## Calculate scores for representative terms
## and plot heat map using term descriptions
representative_df <- example_pathfindR_output_clustered[example_pathfindR_output_clustered$Status == "Representative", ]
score_matrix <- score_terms(
enrichment_table = representative_df,
exp_mat = example_experiment_matrix,
cases = cases,
use_description = TRUE, # default FALSE
label_samples = FALSE, # default = TRUE
case_title = "RA", # default = "Case"
control_title = "Healthy", # default = "Control"
low = "#f7797d", # default = "green"
mid = "#fffde4", # default = "black"
high = "#1f4037" # default = "red"
)
Comparison of 2 pathfindR Results
The function combine_pathfindR_results() allows
combination of two pathfindR active-subnetwork-oriented enrichment
analysis results for investigating common and distinct terms between the
groups. Below is an example for comparing results using two different
rheumatoid arthritis-related data
sets(example_pathfindR_output and
example_comparison_output).
combined_df <- combine_pathfindR_results(
result_A = example_pathfindR_output,
result_B = example_comparison_output,
plot_common = FALSE
)
#> You may run `combined_results_graph()` to create visualizations of combined term-gene graphs of selected termsFor more details, see the vignette Comparing Two pathfindR Results
Analysis with Custom Gene Sets
As of v1.5, pathfindR offers utility functions for obtaining organism-specific PIN data and organism-specific gene sets data via
get_pin_file()andget_gene_sets_list(), respectively. See the vignette Obtaining PIN and Gene Sets Data for detailed information on how to gather PIN and gene sets data (for any organism of your choice) for use with pathfindR.
It is possible to use run_pathfindR() with custom gene
sets (including gene sets for non-Homo-sapiens species). Here, we
provide an example application of active-subnetwork-oriented enrichment
analysis of the target genes of two transcription factors.
We first load and prepare the gene sets:
## CREB target genes
CREB_target_genes <- normalizePath(system.file("extdata/CREB.txt", package = "pathfindR"))
CREB_target_genes <- readLines(CREB_target_genes)[-c(1, 2)] # skip the first two lines
## MYC target genes
MYC_target_genes <- normalizePath(system.file("extdata/MYC.txt", package = "pathfindR"))
MYC_target_genes <- readLines(MYC_target_genes)[-c(1, 2)] # skip the first two lines
## Prep for use
custom_genes <- list(TF1 = CREB_target_genes, TF2 = MYC_target_genes)
custom_descriptions <- c(TF1 = "CREB target genes", TF2 = "MYC target genes")We next prepare the example input data frame. Because of the way we choose genes, we expect significant enrichment for MYC targets (40 MYC target genes + 10 CREB target genes). Because this is only an example, we also assign each genes random p-values between 0.001 and 0.05.
set.seed(123)
## Select 40 random genes from MYC gene sets and 10 from CREB gene sets
selected_genes <- sample(MYC_target_genes, 40)
selected_genes <- c(
selected_genes,
sample(CREB_target_genes, 10)
)
## Assign random p value between 0.001 and 0.05 for each selected gene
rand_p_vals <- sample(seq(0.001, 0.05, length.out = 5),
size = length(selected_genes),
replace = TRUE
)
example_pathfindR_input <- data.frame(
Gene_symbol = selected_genes,
p_val = rand_p_vals
)
knitr::kable(head(example_pathfindR_input))| Gene_symbol | p_val |
|---|---|
| HNRNPD | 0.01325 |
| IL1RAPL1 | 0.00100 |
| CD3EAP | 0.00100 |
| LTBR | 0.02550 |
| CGREF1 | 0.00100 |
| TPM2 | 0.05000 |
Finally, we perform active-subnetwork-oriented enrichment analysis
via run_pathfindR() using the custom genes as the gene
sets:
example_custom_genesets_result <- run_pathfindR(
example_pathfindR_input,
gene_sets = "Custom",
custom_genes = custom_genes,
custom_descriptions = custom_descriptions,
min_gset_size = 1, # do not limit the gene set size for demo
max_gset_size = Inf, # do not limit the gene set size for demo
)
knitr::kable(example_custom_genesets_result)| ID | Term_Description | Fold_Enrichment | occurrence | support | lowest_p | highest_p | Up_regulated | Down_regulated |
|---|---|---|---|---|---|---|---|---|
| TF2 | MYC target genes | 18.61497 | 1 | 0.6822917 | 0.0e+00 | 0.0e+00 | ADPRHL1, AEN, ANKRD13B, ATP6V1C1, BMP2, CCNYL1, CFL1, CNNM1, DOHH, FAM76B, FGF11, FHOD1, FLT3, GAPDH, GNAS, GNB2, HEBP2, ILF3, JOSD1, JPH1, KCNQ5, LHX9, MAP7, NEURL2, POP1, PPP1R3C, REEP3, RELB, SLC12A6, SLC39A11, SNTB2, TBL1Y, TIMM9, TMUB1, TSC1, TUBA4A, USP36, ZBTB11 | |
| TF1 | CREB target genes | 20.73646 | 1 | 0.1406250 | 2.7e-06 | 2.7e-06 | CDS1, DHX36, GPBP1, IRF2BPL, MBIP, PPP2R2A, RCAN1, RELB, RUNDC3A, THOC1, ZBTB11 |
It is also possible to run pathfindR using non-human organism annotation. See the vignette pathfindR Analysis for non-Homo-sapiens organisms
Huang DW, Sherman BT, Tan Q, et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8(9):R183.↩︎
Ideker T, Ozier O, Schwikowski B, Siegel AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18 Suppl 1:S233-40.↩︎
Ideker T, Ozier O, Schwikowski B, Siegel AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18 Suppl 1:S233-40.↩︎
Ozisik O, Bakir-Gungor B, Diri B, Sezerman OU. Active Subnetwork GA: A Two Stage Genetic Algorithm Approach to Active Subnetwork Search. Current Bioinformatics. 2017; 12(4):320-8. /1574893611666160527100444↩︎
Huang DW, Sherman BT, Tan Q, et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8(9):R183.↩︎